CytoxPred predictions will be generated by reverse screening based on the similarity principle, which hypothesizes that similar molecules are prone to be toxic for the same cell lines. CytoxPred will be built on a highly curated training set of cytotoxicity phenotypic data extracted from publicly available databases. Importantly and additionally, thanks to our expertise in protein target prediction, we could be in a position to predict not only the cell lines sensitive to given compounds, but also the mode of action of this predicted cytotoxicity, in terms of possible protein binding. This could open the road to drug discovery and development strategies relying not only on existing experimental information but also on robust biological activity predictions. We will focus on cancer-related compounds and cell lines, to benefit from our expertise in Oncology. Experimental validation will be performed by the group of Prof. Elisa Oricchio, EPFL.
Research Projects
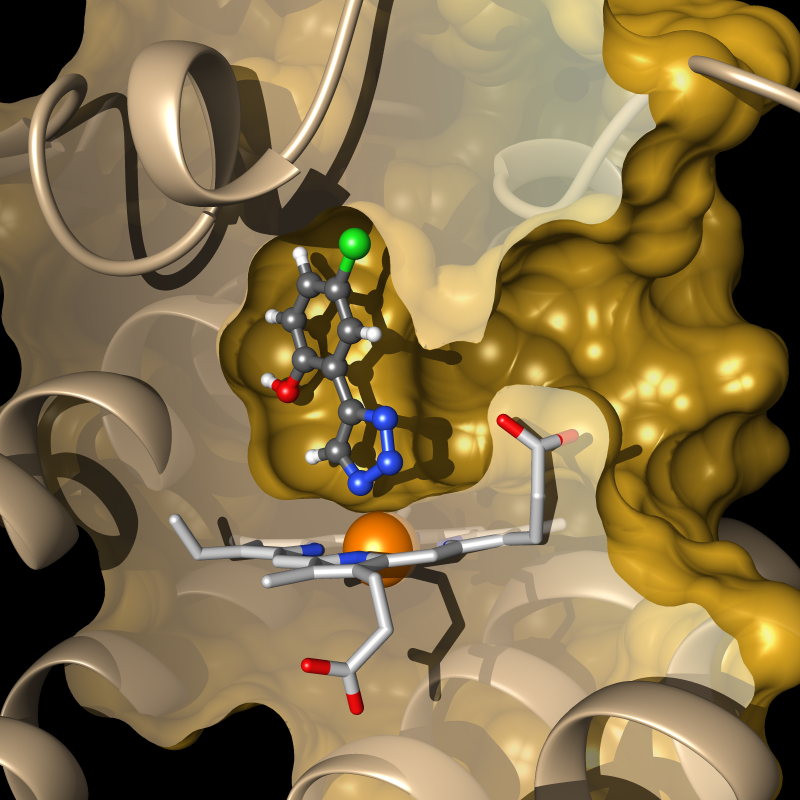
Immuno-oncology has delivered exciting results over the last decade, characterized by substantial and long-term clinical benefit for cancer patients. One promising strategy to further improve clinical outcomes consist in inhibition of the hemoprotein indoleamine 2,3-dioxygenase 1 (IDO1), which plays a major role in tumor-induced immunosuppression. We are rationally designing small molecule modulators of IDO1 using different computational and experimental techniques. Our fragment-based computational drug-design effort led to the discovery of highly efficient IDO1 inhibitors, the most active being of nanomolar potency both in enzymatic and cellular assays, while showing no cellular toxicity and a high selectivity for IDO1 over related enzymes. Several co-crystal structures of our compounds confirm the predicted binding modes and yield insight into the mechanism of IDO1 inhibition.
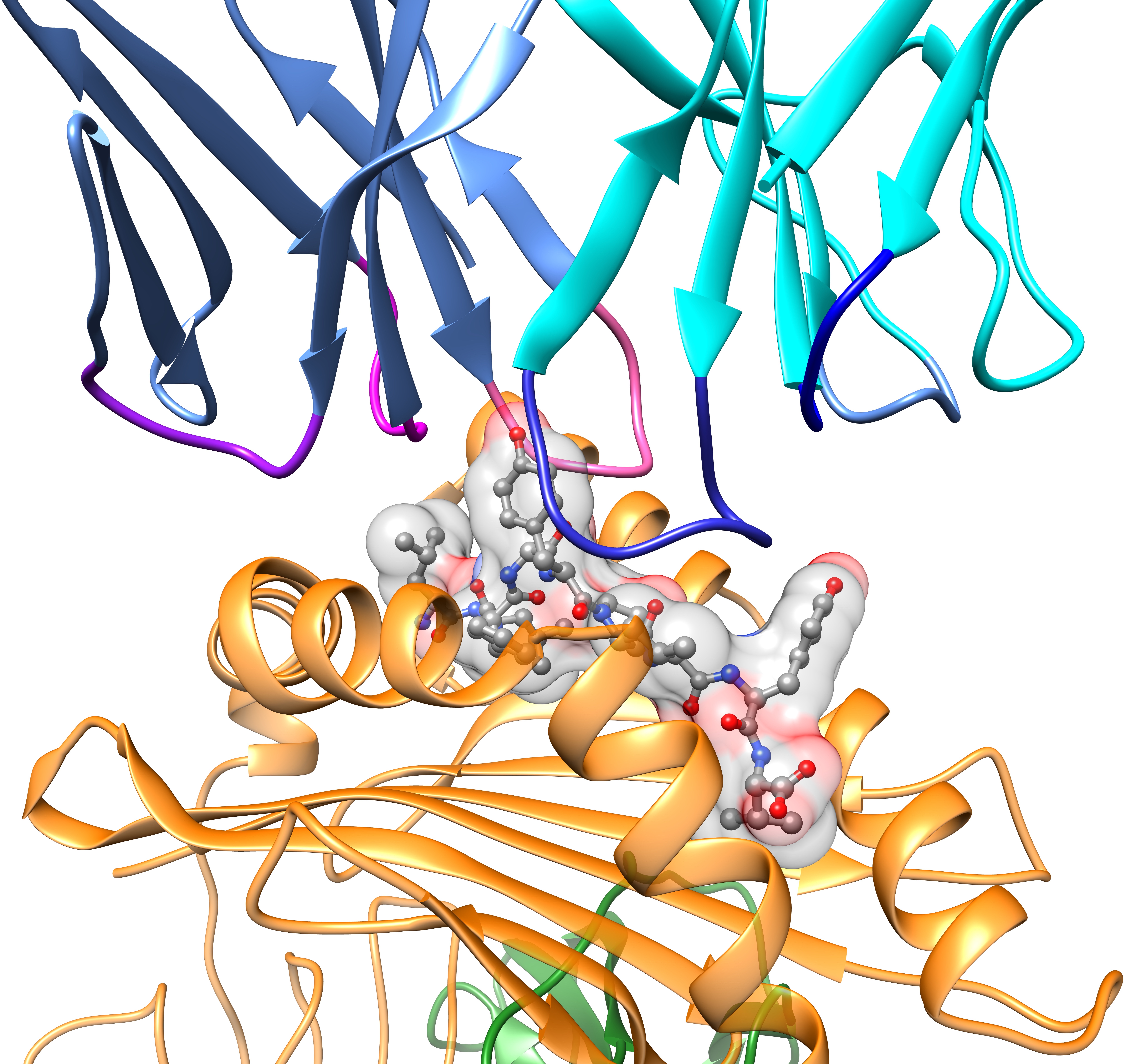
Protein engineering for cancer immunotherapy
We have developed or adapted several free energy calculation methods, including a MM-GBSA approach, for protein engineering. These methods were used to optimize several T-cell receptors (TCR) sequences that play an important role in cancer patients' immune response. Several sequence optimizations were developed for a TCRs recognizing melanoma antigens in order to increase their affinity and specificity. T-cells transduced with this modified gene have shown a greater ability to reproduce and kill antigen-presenting cells. We have also developmed and applied several molecular modelling approaches aiming at predicting TCR/pMHC cross-recognition using structure-based machine learning approaches, predicting the binding mode of peptide epitopes on the MHC molecule, predicting the 3D conformation and binding mode of complementary determining regions of TCR in complex with p-MHC, and finally predicting the exact binding mode orientation of TCR over p-MHC.
Design and application of CytoxPred. Machine-learning to predict cancer cell lines targeted by small molecules
A rapidly growing amount of information regarding phenotypic cytotoxicity of small molecules is currently recorded in publicly available databases, such as ChEMBL. This information has reached a level of diversity, quality and extent that allows its exploitation by machine learning approaches to predict the cell lines that could be sensitive to given small molecules. Capitalizing on our experience in predicting protein targets of bioactive small molecules, our overall goal is to develop CytoxPred, a new and original machine learning model able to accurately predict the cancer cell lines targeted by small molecules.
Attracting Cavities: Towards a QM/MM Algorithm for Docking of Covalent and Noncovalent Ligands
The molecular modelling group of the SIB recently developed a molecular docking code, Attracting Cavities, which first replaces the complex energy landscape of a protein by a grid of attracting points mimicking the target cavities, before reintroducing its actual energy hyper-surface to refine the ligand position. Tested on 85 experimental ligand:protein structures, this method yielded asuccess rate of 80% for reproducing experimental binding modes starting from randomized ligand positions, comparing very favourably to other state-of-the-art docking algorithms. However, as a classical docking code, Attracting Cavities cannot calculate the energetic contributions of covalent bonds. We are therefore developing a hybrid quantum/classical (QM/MM) docking algorithm based onAttracting Cavities to extend its scope to the treatment of covalent ligands. For that purpose, the code will be integrated within a new QM/MM interface, MiMiC, coupling the classical GROMACS code with the DFT-based CPMD code. While computational requirements will make the code impractical for high-throughput virtual screening, it will be valuable for hit-to-lead optimization andfragment-based drug design.
Predicting the impact of uncharacterized mutations on the structure and activity of proteins
We have created the Swiss-PO new web tool to map gene mutations on the 3D structure of corresponding proteins, and assess intuitively the structural implications of protein variants for precision oncology. Swiss-PO is constructed around a manually curated database of 3D structures, variant annotations and sequence alignments, for a list of 50 genes taken from the Ion AmpliSeqTM Custom Cancer Hotspot Panel. The web site was designed to guide users in the choice of the most appropriate structure to analyze regarding the mutated residue, the role of the protein domain it belongs to or the drug that could be selected to treat the patient. The importance of the mutated residue for the structure and activity of the protein can be assessed based on the molecular interactions exchanged with neighbor residues in 3D within the same protein or between different biomacromolecules, its conservation in orthologs, or the known effect of reported mutations in its 3D or sequence-based vicinity. Swiss-PO is available free of charge or login at http://www.swiss-po.ch.
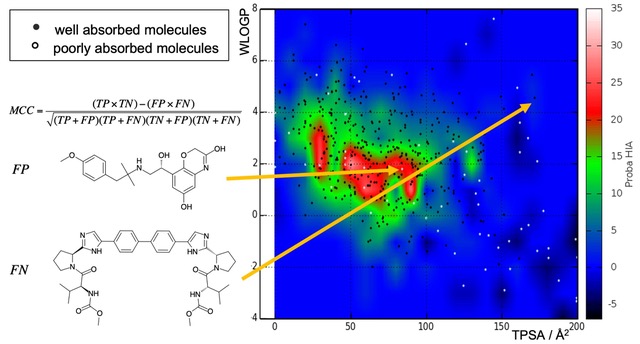
Intending to discover new drugs, compound selection must consider not only intrinsic potency on the protein target but numerous diverse properties. Deficiency in absorption, distribution, metabolism, excretion and toxicity (ADMET), or potential liability in experimental settings because of poor solubility, aggregation behavior, instability are just a few properties to filter out problematic chemical series at early steps and concentrate on the most promising molecules. Since the booming number of molecular structures virtually available, accurate and efficient computational models are needed to predict which molecules to promote further for experimental assays.
Our current developments are focusing on training machine-learning algorithms on tens of thousands of small molecular structures - principally Support Vector Machines and Random Forests - to predict hERG blockade (cardiotoxicity), plasma protein binding (governing distribution) and aggregation (false positives in HTS). These will complement the portfolio of predictors made freely accessible through our SwissADME Web tool.
Adding estimation of affinity to target prediction
Today, prediction of protein target for any bioactive small molecules is obtained through virtual screening and classification models for finding the most similar known active compounds. We are currently performing a broad statistical analysis of bioactivity databases to uncover more quantitative aspect of the similarity principle, which holds that similar molecules are likely to exert similar bioactivities.
Our preliminary results show the existence of a relationship between the quantified molecular similarity of two small molecules and the similarity of affinity on the same protein target. Confirming such effect on large chemical and biological spaces with different measures of similarity (2D and 3D) will foster the development of new machine-learning models able to estimate the level of affinity of small molecules on their predicted probable targets. We expect that such additional bioactivity information would be an important asset for future version of our SwissTargetPrediction reverse screening Web tool.
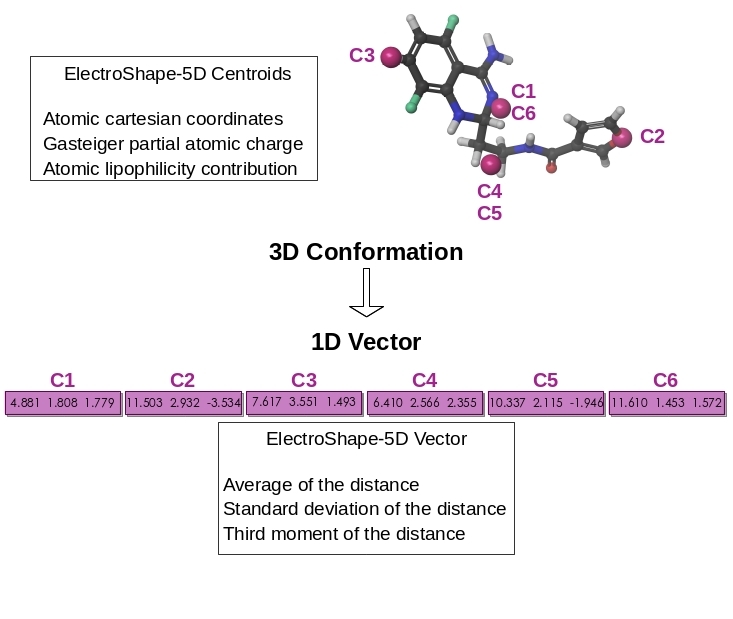
Enhancement of the 3D-shape approach for ligand-based virtual screening
The similarity principle is a rule stating that similar molecules are prone to exhibit similar properties, including biological activity. We have validated and quantified this empirical rule for both 2D- and 3D-similarity measurements, e.g. FP2 fingerprints or ElectroShape.
ElectroShape is a non-superpositional method able to efficiently mine the 3D-shape of molecule into a 1D-vector. Atomic contributions to certain properties has been proven a useful addition to the atomic coordinates for evaluating more accurately the similarity between molecules. Today, Electroshape-5D - shape enriched by projection of electrostatic charge and lipohlicity - is routinely employed in Ligand-based virtual screening (LBVS) since the suitable balance between accuracy and speed. We are currently investigating a rigourous way to addition further 3D-physicochemical properties to the ElectroShape definition. The impact of such novel definition of molecular similarity on the performance of LBVS will be carefully evaluated as well as the implementation in our in-house screening technologies. Also, our Web tools for direct LBVS and for reverse LBVS, SwissSimilarity and SwissTargetPrediction, will greatly benefit from these advances.
Ligand-protein interaction fingerprints for structure-based drug design
Interaction Fingerprints encode information regarding ligand-protein interactions, which is intrinsically tridimensional, into 1D vectors: this allows to compare and analyze the potential binding modes in a faster and more efficient way. In particular, our aim is to develop a procedure that, based on the currently available ligand-protein structures in databases such as the Protein Data Bank, will allow to select the best ligand poses out of those identified by molecular docking software.
This approach has the advantage to evaluate the ligand-protein binding modes in an unbiased way, i.e. considering all types of intermolecular interactions regardless their nature, rather than pre-defined ones only (such as hydrogen bonds).